
Eliminating Target Anopheles Proteins to Non-Target Organisms based on Posterior Probability Algorithm
Volume 6, Issue 1, Page No 710-718, 2021
Author’s Name: Marion Olubunmi Adebiyia), Oludayo Olufolorunsho Olugbara
View Affiliations
ICT and Society Research Group, Luban Workshop, Durban University of Technology, P.O Box 1334, Durban 4000, South Africa
a)Author to whom correspondence should be addressed. E-mail: mariona@dut.ac.za
Adv. Sci. Technol. Eng. Syst. J. 6(1), 710-718 (2021); ![]() DOI: 10.25046/aj060177
DOI: 10.25046/aj060177
Keywords: Alignment, Anopheles, Homology, Organism, Probability

Export Citations
Capturing similarity in gene sequences of a target organism to detect significant regions of comparison will most likely occur because genes share a related descendant. Local sequence alignment for the targeted organisms can help preserve associations among sequences of related organisms. Such homologous genes possess identical sequences with common ancestral genes. The genes may be similar to common traits, and varying purposes, but they descend from a common ancestor. Basic local alignment search tool (BLAST) from the National Center for Biotechnology Information. (NCBI) has been used by different researchers to resolve the various forms of alignment problems. However, much literature to bare the efficacy of standard protein-protein BLAST (BLASTp) on the MATLAB platform has not been seen. In this study, a position-specific iteration BLASTp of 20 anopheles insecticide target protein sequence was performed on NCBI Ensembl against genomes of Anopheles (target organism), then against humans, fruit-fly, zebrafish, and chicken genomes (non-target organisms) to eliminate the targets with homology to non-target organisms. Furthermore, the same iteration was repeated for the genomes of Anopheles and non-target organisms using a posterior probability algorithm built into MATLAB as a tool for protein to protein search BLAST. Outputs from NCBI and MATLAB were put forward to determine the optimality of an optimized search algorithm on MATLAB. The MATLAB-Blastp method based on the application of posterior probability has helped to avoid errors occurring in the early stages of alignment. Moreover, the same results were obtained for the sought features on NCBI Blastp with a refined understanding of how feature values are generated from MATLAB posterior probability built-in algorithm for position-specific BLAST.
Received: 03 September 2020, Accepted: 07 January 2021, Published Online: 05 February 2021
1.Introduction
Local alignment algorithms are suitable for unrelated sequences that are assumed to comprehend areas of comparable sequence motifs within a larger sequence framework [1]. Alignment is a mutual procedure of two sequences exhibiting positions where sequences are similar or dissimilar. A sequence alignment establishes residue-to-residue correspondences among sequences such that the order of residue in each sequence is preserved. Sequence analysis is a subject of deoxyribonucleic acid (DNA), ribonucleic acid (RNA) or peptide sequence for wide variations of analytical techniques to comprehend its purpose, structure, and development [2].
Many dynamic programming algorithms such as Smith-Waterman, FASTA, and BLAST algorithms were developed for accomplishing local alignment. The Smith–Waterman algorithm accomplishes the task of local alignment of sequences for defining comparable neighborhoods of regions between two nucleotides or protein sequences. The algorithm captures the segments of all likely lengths and optimizes a comparative measure, instead of stretching over the entire sequence being process [3]. FASTA program was written for comparing protein sequences but it was later modified to conduct searches on DNA [4]. FASTA software uses the principle of finding similarity between two sequences statistically. This software matches one sequence of DNA or protein with the other by local sequence alignment method. It searches for local region for similarity but not the best match between two sequences. Since this software compares localized similarities at a time, it can come up with a mismatch. FASTA takes a small part of a sequence known as k-tuples where a tuple can be from 1 to 6, matches it with k-tuples of other sequence and once a threshold value of matching is reached it generates result. It is a program that is used to shortlist prospects of matching large sequences because it is very fast. BLAST is frequently used for relating data sequences, recovering, and extracting sequences from databases in bioinformatics [5]. It has shown useful contributions in molecular biology, computational biology, and molecular genetic [6]. BLAST presents reliable and fast statistical reports, flexible search algorithm, and heuristic search methods [7]. BLAST based algorithms seek to extract a snippet of a query sequence that has a perfect alignment with a fragment of a targeted sequence found in a database. In the original BLAST algorithm, the chopped fragment is becoming the input to extend alignment in both query and subject database. BLAST searches for short sequences in an input query that matches short sequences in a database [8]. The nucleotide-nucleotide search, megaBLAST, BLASTN, BLASTP, BLASTX, TBLASTN are other architypes of BLAST algorithms [9].
Dynamic programing through the BLAST algorithm with various variants has made homologous search on genome databases of organisms possible. The aim of such analysis was to predict genes that are homologous in similar or dissimilar species. Gene prediction depends mostly on comparing a genomic sequence with a complementary DNA (cDNA), or protein database. However, most results are inaccurate for several reasons, including incomplete reference databases and lack of contribution to analysis of species. Position specific iterative BLAST (PSI-BLAST) is a program that finds distance relative to a protein. It creates a list of all closely related proteins that were combined into a general “profile” sequence, and summarizes significant features found in protein sequences. A query against a protein database is performed with in-built profile to extract larger group of proteins. The posterior probability is implemented using Bayesian theorem that involves revising a prior profile sequence that is extracted by psi-blast. This algorithm takes into consideration a new sequence profile information, which is a larger profile sequence group used to construct a profile and the process was iterated for four identical non-target organisms. It is believed that PSI-BLAST is much more sensitive in picking up distance based evolutionary relationships than a standard normal protein-protein BLASTP [10].
In this study, a local alignment algorithm has been developed based on BLASTP (protein query sequence against protein database search) and PSI BLAST (for more sensitive protein–protein similarity searches) to determine the effectiveness of the two search algorithms. A literature review to extract one of the best local search algorithms (Smith Waterman algorithm) was conducted. The NCBI BLAST was used to implement a protein query against protein database with a known essential protein sequences with PSI-BLASTP combined with posterior probability to generate an updated list of corresponding homologous protein from four other closely identical organisms. NCBI BLAST is widely used to implement sequence alignment, but very few works have implemented the local BLAST on MATLAB environment. Consequently, we are proposing a distinct way of elucidating homologous genes of a target organism when compared to selected non-target organisms through sequence alignment. This work is relevant for predicting genes that must be targeted in anopheles when formulating new compounds for drug target. If such gene is tampered with in the target anopheles during insecticidal spray, what happens to non-target organisms such as human, fruit flies, chickens, and fishes. There is a high tendency of harming non-targeted organisms during insecticidal spray if homologous genes are not eliminated in the target gene list before insecticidal compounds are formulated and recommended for use. Local sequence alignment for a targeted specie can help preserve associations among sequences of related specie. Such homologous genes possess identical sequences with common ancestral gene and are useful for function prediction and characterization.
2. Related Works
Orthologs are homologous genes that diverge after a speciation event, and still have their main functions conserved. A homologous gene is inherited from two species by a common ancestor and homologous genes can be similar in sequence. But homology is the existence of the same body morphological structures in different organisms. It is an important concept of evolution and comparative biology [11]. The availability of genome sequence of several species has provided an opportunity to elucidate the effects of evolution on every nucleotide and protein in a genome [12-14]. It is easy to identify nucleotide sets that descended from a common ancestral nucleotide with sequence alignment because the problem of identifying an evolutionary related nucleotide and protein is the sequence alignment [15]. Strategies for aligning multiple and entire genomes of organisms include ‘local’ alignment and ‘global’ alignment [13, 15]. Their work has demonstrated a typical example of evolutionary scenario that involved the replication of double-stranded DNA in a parent cell (target organism) and division into two child cells. Their result showed two positions of an undirected duplication because of slippage replication, and occurrence of a directed duplication involving an RNA intermediate.
Studies on efficiency of alignment algorithm for homologous sequence similarity search in a genomic database was performed on a single [3]. The dynamic programming was deployed to isolate similar regions in gene sequences as a form of comparative analysis [16]. The authors discovered a suitable technique to calculate similarity in protein gene sequences within and across related organisms. They proposed a technique that computes sequence similarity, and their algorithm was evaluated using data sets from various species. The ‘best-in-genome’ method has been introduced [17], where a pairwise local alignment [18] between rat (target genome) and human genome (non-target genome) was initiated. The filter kept the best alignment for each position in rat genome and generated many to one relationship between rat genome and human genome but did not capture all orthologous relationships. The result was a reference-based multiple alignment with a property that gave every column, at most one position from each genome. A vertebrate local aligner with a faster nucleotide and more sensitive cross-species protein alignments has been constructed [19]. A web-based BLAST server for human genome makes homologous search possible and serves the purpose multiple genome alignments for yeast, insects, and vertebrates [20]. Aligning human, mouse, and rat genome [21] with progressive extension pairwise alignment orchestrated for human to mouse alignment has been reported [22]. Researches have combined strategies and tools for whole genome alignments [23-27], but none of the previous works have been found to specifically provide the Psi-Blastp by Bayes theorem posterior probability for generating homologous alignment of anopheles to human, fruit-fly, zebra-fish and chicken genomes as provided in this work.
3. Experimental
This work has been implemented using minimum hardware and software requirements of a computer system with at least 16GB of RAM, 1TB hard disk capacity, Intel Core i7 Microprocessor, with VGA monitor compatible of at least 640/480 resolution and enhanced keyboard with a mouse. The software requires windows operating 7, or higher version, a Blosum62, BLAST standalone database, MATLAB R2016B, and online Ensembl NCBI Database [28] and [29]. The 20 essential genes identified to be potential insecticidal targets for malaria vector were used as input data for Mosquito Anopheles Gambiae [30]. The original data for analysis were extracted from Kyoto Encyclopedia of Genes and Genomes (KEGG) database and AnoCyc database on BioCyc, https://biocyc.org/organism-summary?object=ANO [31]. The detailed summary of Anopheles gambiae, version 24.1 KEGG described a collection of databases dealing with genomes, biological pathways, diseases, drugs, and chemical substances. BioCyc is a swarm of about 5700 pathway/genome databases (PGDBs) specific to various organisms. Each PGDB houses a predicted metabolic network and full genome of a specific organism, including reactions, metabolites, metabolic pathways, enzymes, proteins, genes, and lots of other components [32]. The protein sequence of these genes was extracted from protein database at NCBI as flat file or FASTA format from Genebank. Table 1 shows the dataset features of twenty genes as enzyme name, protein name, gene name, enzyme commission (EC) number and gene identity (ID).
The implementation of a basic local alignment method using a query against protein database was performed in this study using posterior probability function in MATLAB. The goal was to ascertain the creation of position specific score profile matrix from an alignment. The BLASTp program in MATLAB Bioinformatic tool was designed to map sequences of 20 previously identified Anopheles gambiae insecticidal target genes unto all available protein sequence databases of a specified organism, disease, population, or proteome. Bayesian posterior probability was calculated from psi-blast output to capture the related proteins from distance species during its search. This has resulted into a larger profile sequence group used to construct the final profile information. The process was repeated for the four non-target organisms investigated in this study.
Table 1: Dataset Features
| S/N | AnoCyc (BioCyc) Enzyme Name | Uniprot Protein Name | Gene Name (Uniprot) | EC Number | Gene ID |
| 1 | Alkyl hydroperoxide reductase subunit C Thiol specific antioxidant | Thioredoxin-dependent peroxidase | TPX1 | 1.11.1.15 | AGAP000396 |
| 2 | Cytochrome P450 B-class | AGAP012295-PA | CYP9L1 | 1.14.14.1 | AGAP012295 |
| 3 | Cytochrome P450 | AGAP002429-PA | CYP314A1 | 1.14.99.22 | AGAP002429 |
| 4 | Betaine aldehyde dehydrogenase | AGAP003578-PA | 1274242 | 1.2.1.3 | AGAP003578 |
| 5 | Ribosomal RNA adenine dimethylase | rRNA adenine N (6)-methyltransferase | 1274612 | 2.1.1.183 | AGAP004465 |
| 6 | Methyltransferase type 11 | 2-methoxy-6-polyprenyl-1,4-benzoquinol methylase, mitochondrial | coq5 | 2.1.1.201 | AGAP010488 |
| 7 | tRNA (guanine9-N1)-methyltransferase | tRNA methyltransferase 10 homolog A | 1271937 | 2.1.1.221 | AGAP000324 |
| 8 | MT-A70-like | AGAP002895-PA | 1273072 | 2.1.1.62 | AGAP002895 |
| 9 | Cholineethanolamine kinase | AGAP000010-PA | 1272266 | 2.7.1.82 | AGAP000010 |
| 10 | Diacylglycerol kinase catalytic domain | Sphingosine kinase | 1270104 | 2.7.1.91 | AGAP006995 |
| 11 | Zn (II)-responsive transcriptional regulator | Phenylalanyl-tRNA synthetase beta subunit | 1274174 | 6.1.1.20 | AGAP003517 |
| 12 | Galactose-binding domain-like | Beta-galactosidase | 1281056 | 3.2.1.23 | AGAP002055 |
| 13 | Carbon-nitrogen hydrolase | AGAP012662-PA | 1269132 | 3.5.1.3 | AGAP012662 |
| 14 | Formylmethionine deformylase | Peptide deformylase | 1271597 | 3.5.1.88 | AGAP003861 |
| 15 | Threonyl-tRNA synthetase class IIa | Prolyl-tRNA synthetase | 1274253 | 6.1.1.15 | AGAP003589 |
| 16 | Leucyl-tRNA synthetase | Leucyl-tRNA synthetase | 1277687 | 6.1.1.4 | AGAP008297 |
| 17 | Ribosomal RNA methyltransferase Spb1 C-terminal | 23S rRNA (uridine2552-2′-O)-methyltransferase | 1274000 | 2.1.1.166 | AGAP004177 |
| 18 | Farnesyl diphosphate synthase | Polyprenyl synthetase | 1269998 | 2.5.1.1 | AGAP007104 |
| 19 | Glucosaminegalactosamine-6-phosphate isomerase | 6-phosphogluconolactonase | 1271093 | 3.1.1.31 | AGAP010866 |
| 20 | FAD-binding type 2 | Alkylglycerone-phosphate synthase | 1274507 | 2.5.1.26 | AGAP004358 |
Block substitution matrix (BLOSUM), which is the default matrix for a protein BLAST algorithm is a substitution matrix used for sequence alignment of protein. The matrix is based on local alignment. Based on block comparisons of sequence from database blocks, it contains multiple aligned un-gapped segments that correspond to the top-most conserved protein regions. The goals are to identify “biologically significant” patterns in protein families by emphasizing regions that are thought to be important to protein function. To look for good “discriminators” that emphasize and identify known family members, while excluding known non-members and to pro-site patterns of “motifs”. The standalone BLAST is a suit of programs that were designed to mimic the NCBI BLAST server, and include “blastall”,” megablast”, and “blastp” that exist in NCBI BLAST suit. Its ease of use and user friendliness features are strong inspiration for its application in this study.
4. Results and Discussion
The protein sequences for each of the 20 insecticidal target genes was blasted against protein databases of four non-target organisms. The organisms are Homo Sapiens (Taxonomy ID: 9609) – human genome, Drosophila (Taxonomy ID: 7227) – fruit fly genome, Danio Rerio (Taxonomy ID:7955) – Zebra fish genome and Gallus Gallus (Taxonomy ID: 9031) – chicken genome. The selection procedure certifies that homologous gene of target organism (anopheles) exists in these four non-target organisms. There is possible homology match, which was identified and should be catered for in case of gene inhibition during insecticidal development. In fact, about 30 sequences were blasted against the whole genome per single iteration. The BLAST was matched with parameters before commencing on further BLASTP commands. The BLAST of proteins was run against the protein database of four targeted organisms. The BLASTP algorithm was implemented on Blosum62 Ensembl database section. During this process we were looking for homologs, which is a measure of relationship between two genes that descended from a common ancestral protein. Their various e-values and percentage identities were identified as the unique selection criteria. It confirmed the literature [33] that homologous genes can be predicted and validated by sequence alignment method.
The e-value measures level of a likelihood that any match in sequences is purely by chance. Consequently, a lower e-value determines the less significant matches made but gives an idea of potential relations among query organisms and database organisms. This is a result of random chance and therefore the most significant gene matches in those non-target organisms were selected as the homologous. Selecting a homolog like the target sequence analyzed, the match with the lowest e-value and highest percentage was classified a significant match or hit. The e-value threshold was set at 0.00001 as a standard with NCBI Ensembl database that was deployed, while the position specific iteration BLAST threshold was set at 0.005. Figures 1 and 2 represent two sample result pages of 20-protein sequences blasted against the four non-target genome databases investigated with e-values and IDs of its homologous gene.
Different values were collected for various organisms blasted on ensembl platform. These values are the homolog, e-value, and percentage identity. Tables 2 and 3 show results of human and zebra fish homologous genes of the four organisms blasted in this study. The extracted database files from the Ftp site for the Anopheles was extracted from ftp://ftp.ncbi.nlm.nih.gov/genomes/ and available on Figure 3 shows this result for the case of human genomic information. The figure constitutes the precise data files and human genome information from NCBI genome databases and similar results were obtained for Zebrafish, Fruit-fly, and Chicken. The databases from the Ftp site were downloaded as GenBank files and converted to Microsoft (MS) Access database. The MS access file was linked as input to the MATLAB for implementation. BLASTP search was performed using MATLAB. A position specific iteration blast for protein query against protein (PSI–BLASTP) was completed for each sequence of anopheles against non-target organism (Human, Zebrafish, Fruit-fly, and Chicken) genome database. This was done to eliminate targets with homolog to non-target organism.
 Figure 1: BLAST result page on ensembl.org
Figure 1: BLAST result page on ensembl.org
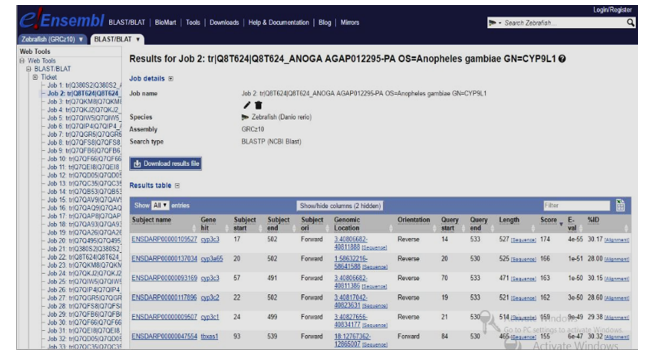 Figure 2: Extracted result with e-value and ID of its homologous gene
Figure 2: Extracted result with e-value and ID of its homologous gene
Finally, 20 anopheles protein targets were screened with two target genomes of Homo sapiens and Drosophila melanogaster. This was to identify isoforms from two organisms when screened with genomes of anopheles. The result of anopheles protein isoforms when screened with homo sapient and drosophila genomes is presented in Table 4. These isoforms can formulate a potential class of protein and can elucidate more molecular and functional variations ciphered in the genome by further functional analysis.
The search for protein isoform in this study may be out of scope because we do not have plans to conduct clinical or computational proteomics analysis and transcriptomic analysis. However, protein isoform is seen as the same protein existing in many different forms, rather it is a new class of protein that may be useful as biomarkers for early diagnostic of clinical proteomics [34]. Studies have shown that traditional methods of protein isoform determination have proved that isoforms can only be determined quantitatively at the transcript level, not in the protein level. Moreover, that condition came with several other disadvantages, high throughput analysis has made it possible, but the data used in this study are not confirmed transcriptomic dataset.
Table 2: Human (homo sapient) homologous gene
| S/N | AnoCyc (BioCyc) Enzyme Name | EC Number | KEGG Gene ID | Homolog Gene ID | E-value | % ID |
| 1 | Alkyl hydroperoxide reductase subunit C Thiol specific antioxidant | 1.11.1.15 | AGAP000396 | ENSP00000389047 | 1.00E-64 | 66.27 |
| 2 | Cytochrome P450 B-class | 1.14.14.1 | AGAP012295 | ENSP00000228606 | 1.00E-12 | 31.63 |
| 3 | Cytochrome P450 | 1.14.99.22 | AGAP002429 | ENSP00000368079 | 1.00E-18 | 23.11 |
| 4 | Betaine aldehyde dehydrogenase | 1.2.1.3 | AGAP003578 | ENSP00000438296 | 1.00E-157 | 57.73 |
| 5 | Ribosomal RNA adenine dimethylase | 2.1.1.183 | AGAP004465 | ENSP00000421754 | 1.00E-107 | 77.91 |
| 6 | Methyltransferase type 11 | 2.1.1.201 | AGAP010488 | ENSP00000449933 | 3.00E-11 | 58.82 |
| 7 | tRNA (guanine9-N1)-methyltransferase | 2.1.1.221 | AGAP000324 | ENSP00000423628 | 1.00E-14 | 35.77 |
| 8 | MT-A70-like | 2.1.1.348 | AGAP002895 | ENSP00000440598 | 1.00E-43 | 34.93 |
| 9 | Cholineethanolamine kinase | 2.7.1.82 | AGAP000010 | ENSP00000398091 | 1.00E-37 | 47.57 |
| 10 | Diacylglycerol kinase catalytic domain | 2.7.1.91 | AGAP006995 | ENSP00000471180 | 1.00E-14 | 38.25 |
| 11 | Zn(II)-responsive transcriptional regulator | 6.1.1.20 | AGAP003517 | ENSP00000367498 | 1.00E-20 | 30.49 |
| 12 | Galactose-binding domain-like | 3.2.1.23 | AGAP002055 | ENSP00000407365 | 1.00E-06 | 46.94 |
| 13 | Carbon-nitrogen hydrolase | 3.5.1.3 | AGAP012662 | ENSP00000356986 | 1.00E-37 | 33.21 |
| 14 | Formylmethionine deformylase | 3.5.1.88 | AGAP003861 | ENSP00000288022 | 1.00E-40 | 39.35 |
| 15 | Threonyl-tRNA synthetase class IIa | 6.1.1.15 | AGAP003589 | ENSP00000358060 | 1.00E-05 | 22.77 |
| 16 | Leucyl-tRNA synthetase | 6.1.1.4 | AGAP008297 | ENSP00000447763 | 1.00E-07 | 21.05 |
| 17 | Ribosomal RNA methyltransferase Spb1 C-terminal | 2.1.1.166 | AGAP004177 | ENSP00000384423 | 1.00E-22 | 37.41 |
| 18 | Farnesyl diphosphate synthase | 2.5.1.1 | AGAP007104 | ENSP00000417865 | 1.00E-05 | 27.04 |
| 19 | Glucosaminegalactosamine-6-phosphate isomerase | 3.1.1.31 | AGAP010866 | ENSP00000471446 | 2.00E-11 | 37.63 |
| 20 | 2.5.1.26 | AGAP004358 | ENSP00000417011 | 1.00E-11 | 25.58 |
Table 3: Zebra fish (danio rerio) homologous gene
| S/N | AnoCyc (BioCyc) Enzyme Name | EC Number | Gene ID | Homolog Gene | E-value | % ID |
| 1 | Alkyl hydroperoxide reductase subunit C Thiol specific antioxidant | 1.11.1.15 | AGAP000396 | ENSDARP00000120934 | 1.00E-49 | 72.5 |
| 2 | Cytochrome P450 B-class | 1.14.14.1 | AGAP012295 | ENSDARP00000122647 | 1.00E-06 | 34.07 |
| 3 | Cytochrome P450 | 1.14.99.22 | AGAP002429 | ENSDARP00000091260 | 1.00E-19 | 24.52 |
| 4 | Betaine aldehyde dehydrogenase | 1.2.1.3 | AGAP003578 | ENSDARP00000012767 | 1.00E-154 | 57.91 |
| 5 | Ribosomal RNA adenine dimethylase | 2.1.1.183 | AGAP004465 | ENSDARP00000124704 | 1.00E-04 | 30.69 |
| 6 | Methyltransferase type 11 | 2.1.1.201 | AGAP010488 | ENSDARP00000131342 | 1.00E-04 | 25.22 |
| 7 | tRNA (guanine9-N1)-methyltransferase | 2.1.1.221 | AGAP000324 | ENSDARP00000109893 | 1.00E-36 | 40.41 |
| 8 | MT-A70-like | 2.1.1.348 | AGAP002895 | ENSDARP00000022188 | 4.00E-154 | 48.34 |
| 9 | Cholineethanolamine kinase | 2.7.1.82 | AGAP000010 | ENSDARP00000019763 | 2.00E-93 | 46.37 |
| 10 | Diacylglycerol kinase catalytic domain | 2.7.1.91 | AGAP006995 | ENSDARP00000117613 | 1.00E-07 | 33.33 |
| 11 | Zn(II)-responsive transcriptional regulator | 6.1.1.20 | AGAP003517 | ENSDARP00000069614 | 4.00E-17 | 31.07 |
| 12 | Galactose-binding domain-like | 3.2.1.23 | AGAP002055 | ENSDARP00000047190 | 1.00E-120 | 38.75 |
| 13 | Carbon-nitrogen hydrolase | 3.5.1.3 | AGAP012662 | ENSDARP00000121828 | 1.00E-69 | 44.09 |
| 14 | Formylmethionine deformylase | 3.5.1.88 | AGAP003861 | ENSDARP00000013949 | 3.00E-43 | 40 |
| 15 | Threonyl-tRNA synthetase class IIa | 6.1.1.15 | AGAP003589 | ENSDARP00000103726 | 1.00E-04 | 22.57 |
| 16 | Leucyl-tRNA synthetase | 6.1.1.4 | AGAP008297 | ENSDARP00000106738 | 5.00E-127 | 63.76 |
| 17 | Ribosomal RNA methyltransferase Spb1 C-terminal | 2.1.1.166 | AGAP004177 | ENSDARP00000138990 | 1.00E-15 | 27.8 |
| 18 | Farnesyl diphosphate synthase | 2.5.1.1 | AGAP007104 | ENSDARP00000059927 | 1.00E-74 | 44.48 |
| 19 | Glucosaminegalactosamine-6-phosphate isomerase | 3.1.1.31 | AGAP010866 | ENSDARP00000124115 | 2.00E-13 | 29.92 |
| 20 | 2.5.1.26 | AGAP004358 | ENSDARP00000136279 | 1.00E-177 | 52.42 |
 Figure 3: Database file genome information for Humans (Homo Sapiens)
Figure 3: Database file genome information for Humans (Homo Sapiens)
Table 4: Anopheles protein isoforms when screened against homo sapient and drosophila genomes
| Anopheles Gene ID | Number of Isoforms for Anopheles | Human Homolog/ Uniprot Gene ID | Number of Isoforms for Human homolog gene | Drosophila Homolog/ Uniprot Gene ID | Number of Isoforms for Drosophila homolog gene |
| AGAP000396 | 0 | ENSP00000389047/ PRDX1 | 3 | FBpp0082927/ Prx3 | 0 |
| AGAP012295 | 0 | ENSP00000228606/ CYP27B1 | 3 | FBpp0088127/ Cyp9b1 | 0 |
| AGAP002429 | 0 | ENSP00000368079/ CYP4V2 | 2 | FBpp0088437/ Cyp12d1-p | 2 |
| AGAP003578 | 0 | ENSP00000438296/ ALDH1A2 | 10 | FBpp0079406/ Aldh | 2 |
| AGAP004465 | 0 | ENSP00000421754/ DIMT1 | 4 | FBpp0291478/ mtTFB1 | 0 |
| AGAP010488 | 0 | ENSP00000449933/ COQ5 | 6 | FBpp0073568/ Coq5 | 2 |
| AGAP000325 | 2 | ENSP00000423628/ TRMT10A | 3 | FBpp0077043/ trmt10a | 0 |
| AGAP002895 | 0 |
ENSP00000440598/ METTL3 |
6 | FBpp0079219/ Mettl14 | 0 |
| AGAP000010 | 0 | ENSP00000398091/ ETNK2 | 7 | FBpp0310123/ eas | 5 |
| AGAP006995 | 0 | ENSP00000471180/ SPHK2 | 8 | FBpp0073346/ Sk1 | 2 |
| AGAP003517 | 2 | ENSP00000367498/ LRRC47 | 2 | FBpp0084021/ beta-PheRS | 0 |
| AGAP002055 | 0 | ENSP00000407365/ GLB1 | 8 | FBpp0078861/ Gal | 2 |
| AGAP012662 | 0 |
ENSP00000356986/ NIT1 |
5 | FBpp0081507/ Dmel\CG8132 | 0 |
| AGAP003861 | 0 | ENSP00000288022/ PDF | 0 | FBpp0081794/ Dmel\CG31373 | 0 |
| AGAP003589 | 0 | ENSP00000358060/ TARS2 | 6 | FBpp0072825/ ProRS-m | 2 |
| AGAP008297 | 0 | ENSP00000447763/ VARS2 | 13 | FBpp0291534/ IleRS-m | 0 |
| AGAP004177 | 0 | ENSP00000364423/ PTCH1 | 12 | FBpp0082832/ CG5220 | 0 |
| AGAP007104 | 0 | ENSP00000417865/ GGPS1 | 3 | FBpp0087266/ Fpps | 0 |
| AGAP010866 | 0 | ENSP00000471446/ PGLS | 4 | FBpp0297905/ Oscillin | 4 |
| AGAP004358 | 0 | ENSP00000417011/ LDHD | 3 | FBpp0070365/ D2hgdh | 3 |
5. Conclusion
In this study, BLAST which is a frequently used to determine sequence similarity by querying various sequences type against databases of various datatypes was experimented. BLAST with 20 Anopheles protein sequence was queried against four non-target organism databases. The results of this study have revealed the same e-values, which indicates that there is no significant homology (all e–values > 0.001) between the data on Anopheles when compared to humans, fruit-fly, zebrafish, and chicken databases. Posterior probability in MATLAB was used for experimentation in this study. The study results have shown obvious insecticidal targets in Anopheles gambiae with no significant homology to humans, fruit-fly, zebrafish, and chicken. Further analysis like synthesizing these targets in an experimental scenario can help close dangling ends in search for new insecticide compounds. This may be a useful endeavor for future research.
6. Data Availability Statement
The original source of data for this study is from a standard and structured public repository KEGG and AnoCyc on BioCyc, (https://biocyc.org/organism-summary?object=ANO). Protein sequence of these genes was extracted from the protein database at NCBI as flat file or FASTA format from Genebank. In addition, contents were extracted from UniProt Knowledgebase @ UniProtKB (https://www.uniprot.org/database/DB-0023). This represents some details from Cross-referenced databases on UniProtKB, Protein knowledgebase and UniParc Sequence archive. This is because our analysis involved fetching data from various public databases to generate outputs as the algorithm required in pursuit of research objective. Links to all data supporting the conclusions of this study is publicly available as indicated by web links.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
The authors would like to appreciate the postgraduate research directorate for postdoctoral sponsorship to the first author.
- V. G. Tumanyan, V. O. Polyanovsky, and M. A. Roytberg, “Comparative analysis of the quality of a global algorithm and a local algorithm for alignment of two sequences,” Algorithms for Molecular Biology, 6(25), 1748-7188, 2011, https://doi.org/10.1186/1748-7188-6-25.
- E. S. Donkor, N. T. K. D. Dayie, T. K. Adiku. “Bioinformatics with Basic Local Alignment Search Tool (BLAST) and Fast Alignment (FASTA)”, Journal of Bioinformatics and Sequence Analysis, 6(1), 1-6, 2014, https://doi.org/10.5897/IJBC2013.0086.
- T. O. Oladele, O. M. Bamigbola, and C. O. Bewaji, “On Efficiency of Sequence Alignment Algorithms,” African Scientist, 10(1), 9-14, 2009.
- D. J. Lipman, and W. R. Pearson, “Rapid and sensitive protein similarity searches”, Science, 227(4693), 1435-1441, 1985, https://doi.org/10.1126/science.2983426.
- C. A. Kerfeld, K. M. Scott. “Using BLAST to teach “E-value-tionary Concepts”, PLoS Biology, 9(2), 1-11, 2011.
- G. G. Syngai, P. Barman, R. Bharali, and S. Dey, “BLAST: An Introductory Tool for Students to Bioinformatics Applications”, Keanean Journal of Science, 2, 67-76, 2013.
- R. S. Neumann, S. Kumar, and K. Shalchian-Tabrizi, “BLAST output visualization in the new sequencing era”, Briefings in Bioinformatics, 15(4), 484-503, 2014, doi: 10.1093/bib/bbt009.
- D. W. Kim, N. R. Kim, D. S. Kim, S. H. Choi, S. H. Chae, H. S. Park, “easySEARCH: A user-friendly bioinformatics program that enables BLAST searching with massive number of query sequences”, Bioinformation, 8(16), 792–794, 2012, doi: 10.6026/97320630008792.
- M. Thomas. “The BLAST Sequence Analysis Tool”, The NCBI Handbook, 2013.
- G. L, Rosen, R. Polikar, R., D. A. Caseiro, S. D. Essinger, B. A. Sokhansanj, “Discovering the unknown: Improving detection of novel species and genera from short reads”, Journal of Biomedicine and Biotechnology, 2011, https://doi.org/10.1155/2011/495849.
- B. K. Hall, “Homology and embryonic development”, In M. K. Hecht, R. J. Macintyre, M. T. Clegg, eds, Evolutionary Biology, Springer, Boston MA, 28, 1–37, 1995, https://doi.org/10.1007/978-1-4615-1847-1_1.
- D. S. Moore “Importing the homology concept from biology into developmental psychology”, Developmental Psychobiology, 55(1), 13–21, 2013, https://doi.org/10.1002/dev.21015.
- I. Brigandt, “Essay: Homology”, In: The Embryo Project Encyclopedia, ISSN: 1940-5030, 2011, https://embryo.asu.edu/view/embryo:124921.
- R. W. Scotland, “Deep homology: a view from systematics”, BioEssays, 32(5), 438–449, 2010, doi: 10.1002/bies.200900175.
- N. D. Colin, P. Lior, “Evolution at the nucleotide level: the problem of multiple whole-genome alignment”, Human Molecular Genetics, 15(1), R51–R56, 2006, https://doi.org/10.1093/hmg/ddl056.
- B. Shankar, D. Vinod, M. Basavaraj, and P. Manjunath. “Comparative analysis of dynamic programing algorithms to find similarity in gene sequences”, International Journal of Research in Engineering and Technology, 2(8), 312-316, 2013.
- W. J. Kent, R. Baertsch, A. Hinrichs, W. Miller, and D. Haussler, “Evolution’s cauldron: duplication, deletion, and rearrangement in the mouse and human genomes”, Proc. Natl Acad. Sci. USA, 100(20), 11484 – 11489, 2003, doi: 10.1073/pnas.1932072100.
- S. Batzoglou, “The many faces of sequence alignment”. Briefings in Bioinformatics, 6(1), 6–22, 2005, doi: 10.1093/bib/6.1.6.
- W. J. Kent, “BLAT—the BLAST-like alignment tool”, Genome Res., 12(4), 656–664, 2002, doi: 10.1101/gr.229202.
- D. Karolchik, R. Baertsch, M. Diekhans, T. S. Furey, A. Hinrichs, Y. T. Lu, K.M. Roskin, M. Schwartz, C.W. Sugnet, D. J. Thomas, et al., “The UCSC Genome Browser Database”, Nucleic Acids Research, 31(1), 51–54, 2003, https://doi.org/10.1093/nar/gkg129.
- M. Brudno, A. Poliakov, A. Salamov, G. Cooper, A. Sidow, E. Rubin, V. Solovyev, S. Batzoglou, and I. Dubchak, “Automated whole-genome multiple alignment of rat, mouse, and human”, Genome Res., 14(4), 685–692, 2004, doi: 10.1101/gr.2067704.
- O. Couronne, A. Poliakov, N. Bray, T. Ishkhanov, D. Ryaboy, E. Rubin, L. Pachter, and I. Dubchak, “Strategies and tools for whole-genome alignments”, Genome Research, 13(1), 73–80, 2003, doi: 10.1101/gr.762503.
- M. Brudno, C. Do, G. Cooper, M. Kim, E. Davydov, E. Green, A. Sidow, S. Batzoglou, “LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA”, Genome Res., 13(4), 721–731, 2003, doi: 10.1101/gr.926603.
- S. Zhao, J. Shetty, L. Hou, A. Delcher, B. Zhu, K. Osoegawa, P. de Jong, W. Nierman, R. Strausberg, and C. Fraser, “Human, mouse, and rat genome large-scale rearrangements: stability versus speciation”, Genome Res., 14(10a), 1851–1860, 2004, doi: 10.1101/gr.2663304.
- A. Siepel, G. Bejerano, J. Pedersen, A. Hinrichs, M. Hou, K. Rosenbloom, H. Clawson, J. Spieth, L. Hillier, S. Richards, G.M Weinstock, R. K Wilson, R. A Gibbs, W. J Kent, W. Miller, D. Haussler, “Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes”, Genome Res., 15(8), 1034–1050, 2005, doi: 10.1101/gr.3715005.
- E. H. Margulies, M. Blanchette, D. Haussler, E. D. Green, “Identification and characterization of multi-species conserved sequences”, Genome Res., 13(12), 2507–2518, 2003, doi: 10.1101/gr.1602203.
- W. M. Fitch, “Distinguishing homologous from analogous proteins”, Syst. Zool, 19(2), 99-113, 1970, doi.org/10.2307/2412448.
- M. Mathur and Geetika, “Multiple Sequence Alignment Using MATLAB,” International Research Publications House, 3(6), 497-504, 2013.
- S. F. Altschul, T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, D. J. Lipman, “Gapped BLAST and PSI-BLAST: a new generation of protein database search programs”, Nucleic Acids Research, 25(17), 3389– 3402, 1997, doi: 10.1093/nar/25.17.3389.
- M. Adebiyi, J. Oghuan, S. Fatumo, E. Adebiyi, R. Jason, “A Functional Workbench for Anopheles gambiae Micro Array Analysis”, Proceedings – UKSim-AMSS 7th European Modelling Symposium on Computer Modelling and Simulation, 138-143, 2013, doi:10.1109/EMS.2013.24.
- R. Caspi, R. Billington, I. M. Keseler, A. Kothari, M. Krummenacker, P. E. Midford, W. K. Ong, S. Paley, P. Subhraveti, P. D. Karp, “The MetaCyc database of metabolic pathways and enzymes, a 2019 update”, Nucleic Acids Res, 48(D1), 445–453, 2020, doi: 10.1093/nar/gkz862.
- R. A. Holt, G. M. Subramanian, A. Halpern, G. G. Sutton, R. Charlab, D. R. Nusskern, P. Wincker, A. G. Clark, J. M. Ribeiro, S. L. Hoffman et al., “The genome sequence of the malaria mosquito Anopheles gambiae”, Science, 298(5591), 149-159, 2002, doi: 10.1126/science.1076181.
- T. Wiehe, S. Gebauer-Jung, T. Mitchell-Olds, R. Guigo. “SGP-1: prediction and validation of homologous genes based on sequence alignments,” Genome Research, 11(9), 1574-1583, 2001, doi.org/10.1101/gr.177401.
- F. Zhang, and Y. C. Chen, “A method for identifying discriminative isoform-specific peptides for clinical proteomics application”, BMC genomics, 17(S7), 522, 2016, doi: 10.1186/s12864-016-2907-8.