Search for New Potential Breast Cancer Inhibitors (MCF7) Based on Molecular Docking and Biological Assay of Pyrazoline Analogue Compounds
Volume 5, Issue 5, Page No 122-126, 2020
Author’s Name: Jasril1, Hilwan Yuda Teruna1, Ihsan 2, Neni Frimayanti2,a)
View Affiliations
1Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Riau. Kampus Bina Widya, Jl. HR Subantas Km 12.5, Pekanbaru 28293, Indonesia
2Sekolah Tinggi Ilmu Farmasi (STIFAR) Riau, Jalan Kamboja, Pekanbaru, 28293, Indonesia
a)Author to whom correspondence should be addressed. E-mail: nenifrimayanti@gmail.com
Adv. Sci. Technol. Eng. Syst. J. 5(5), 122-126 (2020); ![]() DOI: 10.25046/aj050517
DOI: 10.25046/aj050517
Keywords: pyrazoline, breast cancer, MTT assay, Molecular docking

Export Citations
Cancer is the leading cause of death in world. Currently, there are no approved vaccines to avoid the spreading of this disease. Drug discovery have played important role for discover of new potent drugs that could efficiently and cost-effectively. Pyrazoline analogue compounds known to have good potency as anti-cancer. The aim of this study is to observe the potency of these pyrazoline analogue as anticancer using molecular docking. In this study, ten pyrazoline analogue compounds were docked and tested on MCF7 cell line using MTT assay. Based on docking results, compound PH CN1-4F explored three interations with amino acid residues. These interactions are hydrogen bonding with Arg791, hydrophobic interaction with His790 and van der Waals interaction with Asp810. Biological assay was shown that this compound is also have big potency as breast cancer inhibitor with IC50 value of 61.22 µg/mL. Thus, this strategy can be used to identify new potent drugs as new inhibitor for breast cancer.
Received: 10 July 2020, Accepted: 02 September 2020, Published Online: 09 September 2020
1. Introduction
Breast cancer is begun with development of an abnormal breast cells in men and women. There are some types of cancers such as lung cancer, breast cancer, blood cancer, etc. Currently, breast cancer is one type of cancer that caused of death after lung cancer [1]. Cancer is a very serious problem for human health and this disease grow from the body’s cells, which are characterized by uncontrolled, uncoordinated and unwanted cell division [2].
There are some treatment for breast cancer such as using chemotherapy. Chemotherapy is characterized by targeting the functions of receptor such as ERα (alpha estrogen receptor), PR (progesterone receptor), EGFR (epidermal growth factor receptor) etc. Epidermal growth factor receptor (EGFR) is a receptor of tyrosine kinase that plays an important role in normal physiological and cancer conditions [3]. EGFR can be defined as the first receptor that provided evidence for a relationship between cancer and receptor overexpression.
Wala and co-workers [4] has been tested for biological assay of pyrazoline as inhibitor for microbial, anti-inflammatory, antidepressant, and anticancer effects. Sorrounded the reported of biological assay, it is an urgent to say that pyrazolines are not only acceptable for therapy of some type of cancer but it is also can be used for treat another disease. Some of pyrazoline are also can act as an agents for cancer chemopreventive [5]. Surgery, chemotherapy and radiation therapy are some kind of traditional way for cancer treatment. Exspecially for chemotherapy, the crucial goal of this treatment is to discover the specific target of cancer cells without affecting to the normal cells and also to avoid the side effect. Searching for new agents with highly potential and selective anticancer activities is highly demanded [6].
Drug discovery is very complicated and it is always needed a laborious process. Generally, it will begun with screening of compounds from a large database for then will find out the lead of compounds. It is needed high cost and will spend longer time [7, 8]. Thus, computational approach have created a major impact into drug design process. Calculation will reduce the the cost and also the time for research, in addition, it would also help the rapid development of theraupetic methods [9].
One application of computational approach is using molecular docking is very potent way to discover new potential agent like breast cancer inhibitor [10]. Molecular docking can be applied to determine the binding interaction between the protein and ligand, thus it can estimate the ability of thus compounds (i.e. ligand). The aim of this research is to discover new pyrazoline analogue compounds with good activity for treat breast cancer using molecular docking and approved using biological assay test.
2. Research Methodology
2.1. Receptor 3D Structure
Autodock 1.5.4 software packages was used to perform the molecular docking. Protein data bank (http://www.rscb.org/pdb) was then used to download the protein molecular structure. In this research, the protein tyrosine kinase with PDB ID 1t46 was applied as receptor. It was obtained with resolution of 1.6 Å and also must kept in rigid position. Next step are process to add atoms, merging, checking and repairing missing atoms. This step is begun with added hydrogen atoms, following with merging non-polar hydrogen atoms, checking and repairing missing atoms. For atoms charge, it was started with added Gasteiger charges, following with checking charges and fixing charges on the residue amino acids. Finally, it is assigning atom types to the protein. Autogrid 4 sofware packages with the protein grid box with dimension of 122 x 116 x 120 point along x, y and z axes and centered on the protein for docking. The grid box was settled with grid spacing of 0.35 Ǻ, it also have to default with some atoms such as carbon, hydrogen an nitrogen.
Table 1 Molecular structure of Ligand
| Structure | Struture |
| PH-CN1-4F | PH-CN2-2Cl |
| PF-CN1-2F | PF-CN1-2Cl |
| PF-CN2-3Cl | PF-CN1-3Cl |
|
PH-CN1-2F |
PF-CN1-4Cl |
| PH-CN2-4F |
PH-CN2-4Br |
2.2. Preparation of Ligand
Ligands consisted of ten compounds were depicted in Table 1. ChemBioDraw 13.0 ultra software package was used to draw each molecular structure of these ligands. These ligands must be kept in flexible. Energy minimization was required for each ligand for then the minimized structure were subsequently prepared with detected root of torsion. Flexible ligand with number of torsions were detected using Autodocktools 1.5.4 softwarre packages.
2.3. Docking of Pyrazoline
Autodock 1.5.4. software package was used to contruct the molecular docking. Lamarckian genetic algorithm (LGA) with the local minimization energy was used to search the best poses with the lowest binding energy. This is used to enable modification of the gene population. Docking was then performed with output clustered, it was selected based on the value of the root –mean-square-deviation (RMSD) tolerance pf 2.0 Å. The rank of docking poses were rank used the docking scores. The scoring function in Autodock software was used to predict the binding free energy between a ligand and the molecule receptor. Furthermore, the complex of protein-ligand with the lowest binding free energy is needed for analysis for each docking poses. The molecular visualization for 2D and 3D dimension of those complexes were performed using the Biovia Discovery Studio Visualizer.
2.4. Biological Assay
Biological activity was determined using MTT assay. It was begun with the MCF-7 cells preparation. 96-well plates are provided for seed the cellst with the size of cell density is approximately of 3 x 104 cells cm-3. Incubation was performed for 24 h for cell attachment and growth by adding samples with variation concentration. The next step is dissolving compounds with DMSO based on the required concentration. Subsequently, six desirable concentrations were prepared using PBS (phosphoric buffer solution at pH of 7.30 – 7.65). Different treatment was done for control, it dissolved only with DMSO. The incubation period of 48 h was terminated for the biological assay, it is then added with MTT reagent [3- (4,5-dimethyltiazol-2-yl) -2,5-diphenyl tetrazolium bromide; can also be referred to as thiazole blue] and can incubate continuously for 4 hours. The addition of the MTT reagent should be stopped when a solution containing SDS (sodium dodecyl sulfate) is added, while the required incubation period of 24 hours is also carried out. Microplate reader at 550 nm was used to read the opticacal density. A plot graph that explains the percentage of living cells are compared with the standard (%), it can be used to take the IC50 value and the biological activity value were taken when only PBS and DMSO are accepted, versus the concentration of the tested compound (µM). The IC50 value is the concentration required to inhibit the growth of 50% of infected cells. Each test and analysis was carried out three times and then the values were averaged.The cytotoxic activity of the isolated compounds 1-4 was evaluated against the MCF-7 breast cancer cells according to a method described [11] and Cisplatin (IC50 27.0 µM) was used as a positive control [12].
3. Results and Discussion
3.1. Docking of Pyrazoline
Generally, the main purpose of molecular docking are to identify the binding interaction between a ligand and specific receptor. In addition, it is also to identify the binding pose and the binding free energy. Binding pose is the best pose with the best and selective interaction and orientation between ligand and specific receptor. The scoring fucntion was used to predict and to estimate the binding affinities for then to generate the ligand poses in order to determine the best binding mode. The docking results for these ten pyrazoles are depicted in Table 2.
Table 2: Docking results
| Cpd No | Binding free energy (kcal/mol) | Interaction | ||
| Hydrogen bond | Van der Waals | Hydrophobic | ||
| 1 | -10.98 | Arg791 | Asp810 | His790, Arg791 |
| 2 | -2.01 | Arg791 | His790 | |
| 3 | -5.13 | Arg791 | Glu640, Asp810 | His790 |
| 4 | -9.21 | Arg791 | His790 | |
| 5 | -8.97 | His790 | Asp810 | Arg791 |
| 6 | -8.64 | His790 | Asp810 | Arg791 |
| 7 | -5.47 | Arg791 | Glu640, Asp810 | Arg791 |
| 8 | -3.09 | Arg791 | Glu640 | His790 |
| 9 | -6.79 | Arg791 | His790, Asp810 | |
| 10 | -4.58 | Arg791 | Glu640 | His790 |
The best docking results are selected with some chriteria, they are lowest binding free energy and RMSD (root mean square deviation) less than two. Binding free energy is minimum energy is needed for the ligand to bind with the receptor. RMSD should less than two, it is indicated that the complex of ligand and protein is more stable compare than complex with RMSD is higher than two [13, 14]
According to the docking results, PH CN1-4F was explored some interactions such as hydrogen bonding interaction with Arg791. Van der Waals interaction is also observed with the important amino acid. The hydrogen bonding is constructed between nitrogen of ligand with residue Arg791. Van der Waals interaction was observed with the important residue Asp810. In addition, hydrophobic interaction were also observed with amino acid His790 and Arg791. The computed binding free energy from docking results of -17.65 kcal/mol, it is indicated that the existence of amino acid residue like Arg791, His790 presumably made this compound become active. In addition, the availability of hydrogen bonding, van der Waals and hydrophobic interaction are seem to play an important role in its antagonistic activity [15, 16].
PH CN1-2F, this compound was also observed with one hydrogen bonding. Ligand was bind with Arg791 through hydrogen bonding, hydrophobic interaction was also conducted with amino acid residue His790. Unfortunately, there is no van der Waals interaction between this compound and amino acid. The computed binding free energy value of -10.98 kcal/mol. The absence of van der Waals interaction caused the binding free energy higher than PH-CN1-4F [17]. The spatial arrangement of PH-CN1-4F and PH CN1-2F are depicted in Figure 1.
PF CN2-3Cl and PF CN1-2F were also docked into MCF7 cell line. For PF CN2-3Cl, docking results shown that this compound was able to conduct the hydrogen bonding with Arg791, van der Waals interaction with Glu640 and Asp810. This compound was also able to conduct the hydrophobic interaction with His790, unfortunately this compound has high computed binding energy value of -9.21 kcal/mol. PF CN1-2F was observed hydrogen bonding and hydrophobic interaction with amino acid Arg791 and His790, respectively. The best docking pose for these compounds are depicted in Figure 2.
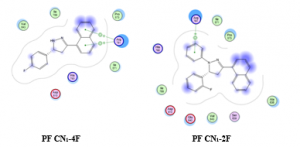
Figure 1: the spatial arrangement for PH CN1-4F and PF CN1-2F
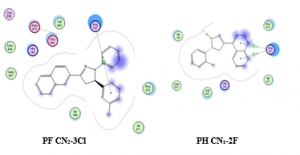
Figure 2: The best docking pose for PF CN2-3Cl and PH CN1-2F
The docking results PH CN2-4F and PH CN2-2Cl were observed with another spatial arrangement like four previous compounds. Hydrogen bonding was exhibited with His790, van der Waals interaction with Asp810 and hydrophobic interaction with Ar791. This seem that those compounds are less active for inhibiting the MCF7 cell line. The spatial arrangement of PH CN2-4F and PH CN2-2Cl are presented in Figure 3.
PF CN1-2Cl showed to have one hydrogen bonds (blue dashed line) with residue Arg791. In addition, van der Waals interaction was also found between ligand and residue Glu640 and Asp810. Residue His790 showed interaction with the ligand through hydrophobic interaction suggesting the importance of this residue in the formation of hydrophobic binding pocket. Likewise, in the case of PF CN1-4Cl, one hydrogen bond was observed between the ligand and residues Arg791. This ligand also displayed van der Waals interaction and hydrophobic interaction via Asp810 and His790, respectively. Best pose for those compounds are presented in Figure 4.
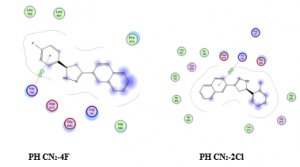
Figure 3. Spatial arrangement of PH CN2-4F dan PH CN2-2Cl
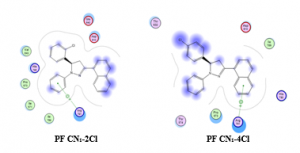
Figure 4. Best pose for PF CN1-2Cl and PF CN1-4Cl
Our results showed PF CN1-3Cl and PF CN2-4Br exhibited Van der Waals interactions with Glu640 and His790. In addition, hydrogen bonding interaction was also observed between these two ligands and the residues Arg791, indicating another possible mode of interaction between these ligands and MCF7 cell line. Existance of van der Waals interaction with Glu640 and His790 indicated that this compound has anoter mode of interaction and it may caused this compund become not active compound. The spatial arrangement of those compounds are depicted in Figure 5.
3.2. Bioasssay of Pyrazoline
Invitro toxicity of pyrazole analogue compounds was determined using MTT assay method against MCF7 cell line. The biological assay for ten pyrazoles are presented in Table 3. Based on Inhibition concentration (IC50), it was observed that there is only one active compounds i.e. PH CN1-4F with IC50 value of 61.22 µg/mL. This data was validated by data from computational calculations (i.e. docking) which is indicated with low value of the binding free energy of this compound. PH CN1-2F, PF CN1-2F, PF CN2-3Cl, PH CN2-4F, PH CN2-2Cl, PF CN1-2Cl, PF CN1-3Cl, PF CN1-4Cl and PH CN2-4Br were shown to have low inhibition activity against MCF7.
PF CN1-3Cl PF CN2-4Br
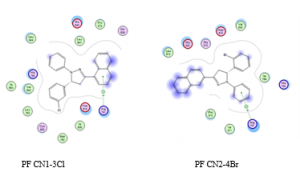
Figure 5. Best pose for PF CN1-3Cl and PF CN2-4Br
Table 3: Biological assay of pyrazoline anologue compounds
| No | Compound | IC50 (µg/mL) |
| 1 | PH CN1-4F | 61.22 |
| 2 | PF CN1-2F | 12739.17 |
| 3 | PF CN2-3Cl | 6575.00 |
| 4 | PH CN1-2F | 99.78 |
| 5 | PH CN2-4F | 123.88 |
| 6 | PH CN2-2Cl | 181.45 |
| 7 | PF CN1-2Cl | 3665.75 |
| 8 | PF CN1-3Cl | 32115.00 |
| 9 | PF CN1-4Cl | 1507.88 |
| 10 | PH CN2-4Br | 3374.75 |
4. Conclusion
Ten pyrazoline analogue compounds were docked and tested on MCF7 cell line using MTT assay. Based on docking results, compound PH CN1-4F explored three interactions with amino acid residues. These interactions are hydrogen bonding with Arg791, hydrophobic interaction with His790 and van der Waals interaction with Asp810. Biological assay was shown that this compound is also have big potency as breast cancer inhibitor with IC50 value of 61.22 µg/mL. Thus, this strategy can be used to identify new potent drugs as new inhibitor for breast cancer.
Conflict of Interest
The authors declare no conflict of interest.
- M.S. Charpentier, R. A. Whipple, M. J. Vitolo, Boggs, J. Slovic, K. N. Tompson, L. Bhandary, S. S Martin. “ Curcumin targets breast cancer steam-like cells with microtentacles that persist in mamospheres and promote reattachment “ Cancer Res, 74(4),1250-1260, 2014. https://doi.org/10.1158/0008-5472.CAN-13-1778.
- C. A. Lipinski, F. Lombardo, B. W. Dominy, and P. J. Feeney. “Experimental and computational approaches to estimate solubility and permeablility in drug:discovery and development setting.” Advanced Drug Delivery Reviews, 46 (1-3), 3-26, 2001. https://doi.org/10.1016/S0169-409X(96)00423-1
- T. Mitsudomi, Y. Yatabe, Y. “Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer”. FEBS, 277, 301- 308, 2010. https://doi.org/ 10.1111/j.1742-4658.2009.07448.x.
- X. Y. Meng, H. X. Zhang, M. Mezei, M. Cui. 2011. “Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery”. Curr Comput Aided Drug Des. 7, 146-157, 2011. https://doi.org/10.2174/157340911795677602.
- S. E. Wala, A. M. Neama, M. K. Emad, K.. Mahmoud, M. M. Mounier. “Synthesis, Biological Evaluation and Docking Analysis of Some Novel Quinazolin Derivatives as Antitumor Agents” Iranian J Pharm Res, 15, 179-196, 2016
- A. Baldi. “Computational Approaches for Drug Design and Discovery: An Overview” Sys Rev Pharm, 1, 1, 99-105, 2010. https://doi.org/10.4103/0975-8453.59519
- Z. Shu-Feng, and Z.Wei-Zhu. “Drug Design and Discovery: Principles and Applications.” Molecules, 22, 279-284, 2017. https://doi.org/ :10.3390/molecules22020279
- S. H. Reich, S. E, Webber. Structure-based drug design (SBDD): Every structure tells a story. Perspectives in Drug Discovery and Design 1, 371–390, 1993. https://doi.org/10.1007/BF02174536
- C. A. Anderson. The Process of Structure-Based Drug Design. Chemistry and Biology, 10, 9, 787-797. https://doi.org/10.1016/j.chembiol.2003.09.002
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, D. J. T. Warren, H. Bokesch, S. Kenney, R. M. Boyd. “New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening” J. Natl. Cancer Inst, 82, 1107-1112, 1990. https://doi. Org/10.1093/jnci/82.13.1107
- Y. E. Hadisaputri, D. Pharm. T. Miyazaki, S. Suzuki, T. Yokobori, T. Kobayashi, N. Tanaka, T. Inose, M. Sohda, H. Kuwano. “ TNFAIP8 Overexpression: Clinical Relevance to Esophageal Squamous Cell Carcinoma”. Ann. Surg. Oncol, 19, S589-S596, 2012. https://doi.org/10.1245/s10434-011-2097-1
- O. Unsal-Tan, T.Tüylü Küçükkılınç,b B. Ayazgök, A. Balkan and K. Ozadali-Saria. “Synthesis, molecular docking, and biological evaluation of novel 2-pyrazoline derivatives as multifunctional agents for the treatment of Alzheimer’s disease”. Medchemcomm., 10, 6, 1018–1026, 2019. https://doi: 10.1039/c9md00030e
- F. Turkan, A. Cetin, P.Taslimi, S. Halide S. K. İ. Gulçin, Synthesis, characterization, molecular docking and biological activities of novel pyrazoline derivatives. Archiv der Pharmazie. Vol. 352, 6, 2019. https://doi.org/10.1002/ardp.201800359
- N. Frimayanti, V. S. Lee, S. M. Zain, H. A. Wahab, N. A. Rahman. “ 2D, 3D QSAR and pharmacophore studies on thiazoline-4-carboxylic acid derivatives as neuraminidase inhibitors in H3N2 influenza virus” Med. Chem. Res. 23, 1447-1453, 2014. https://doi.org/10.1007/s00044-013-0750-x
- N. Frimayanti, B. Iskandar, M. Yaeghoobi, C. H. Han, S. M. Zain, R. Yusof, N. Arahman. “ Docking, synthesis and bioassay studies of imine derivatives as potential inhibitors for dengue Den2 NS2B/NS3 serine protease”. Asian Pac J Trop. Dis. 7(12), 762-766, 2017. https://doi.org/10.12980/apjtd.7.2017D7-177
- M. Kumari, S. Chandra, N. Tiwari, Subbarao. “3D QSAR, pharmacophore and molecular docking studies of known inhibtors and designing of novel inhibitors for M18 aspartyl aminopeptidase of Plasmodium falciparum”. BMC Struc. Biol. 16(12), 1-11, 2016. https://doi.org/ 10.1186/s12900-016-0063-7.
Citations by Dimensions
Citations by PlumX
Google Scholar
Scopus
Crossref Citations
- Jasril Jasril, Neni Frimayanti, Yuana Nurulita, Adel Zamri, Ihsan Ikhtiarudin, Guntur Guntur, "5-(4-Fluorophenyl)-3-(naphthalen-1-yl)-1-phenyl-1H-pyrazole." Molbank, vol. 2021, no. 1, pp. M1197, 2021.
No. of Downloads Per Month
No. of Downloads Per Country