Stability of Ionized Air Using Density Functional Theory (DFT) for High Voltage Engineering
Volume 5, Issue 1, Page No 222-225, 2020
Author’s Name: Fri Murdiya1, Neni Frimayanti2,a)
View Affiliations
1Department of Electrical Engineering Faculty of Engineering Science Universitas Riau Kampus Bina Widya, Jl. HR, Subantas KM 12.5 Simpang Baru Tampan Riau Indonesia
2 Sekolah Tinggi Ilmu Farmasi (STIFAR) Riau, Jalan Kamboja, Pekanbaru, 28293, Indonesia
a)Author to whom correspondence should be addressed. E-mail: neninfrimayamnti@gmail.com
Adv. Sci. Technol. Eng. Syst. J. 5(1), 222-225 (2020); ![]() DOI: 10.25046/aj050128
DOI: 10.25046/aj050128
Keywords: DFT, Ionization, Exitation energy, High voltage, Gas insulation

Export Citations
In high voltage engineering, air is the one insulation gas that can ionized cause by electrical discharge. Air consisted of many gases such as O2, N2, CO2, H2, etc. In order to study the characteristic of the insulation gas, we can apply density functional theory (DFT) to determine gas ionization level such as charge distribution and excitation energy. These data are shown the stability of gas molecule when ionization process occure cause by electrical discharge, ultraviolet light, etc. For application on ozone production by electrical discharge, the oxygen atom can transform to ozone need energy above the stabiity excitation energy of oxygen.
Received: 24 October 2019, Accepted: 25 November 2019, Published Online: 22 January 2020
1. Introduction
In recent year, the high voltage engineering is focused on electrical discharge on the insulation dielectric in the form solid, liquid and gas. Base on our previous research, computational approach can be used to study the air failure in the high voltage which can be utilized to generate ozone from the ionization process of the oxygen atoms [1]. The ionization process has been checked using homo-lumo application. Some gases such as hydrogen, oxygen, nitrogen, carbon dioxide widely used as high voltage insulation. These gases are also used as an ionized gas for application in the high voltage like to produce plasma. This plasma was then used for physics, chemistry and medicine fileds. Recently, laboratory testing was required to determine the electrical strength (E) of that gas [2] or by kinetic modeling of the breakdown process on the basis of a microscopic electron-molecular cross section [3]. These methods are needed much more time and resource intensive and only can be applied to narrow gas.
There are some obvious advantages of the calculation method compared than traditional methods such as saving cost in term of time and effort. Moreover the calculation approach allows for identifying the atomistic processes of insulation gas, and also for exploring the intrinsic changes inside the sensing materials. Along with the development of computational materials science, the theoretical calculation methods can not only be used to provide the reasonable and profound explanations for the experimental results, but they are helpful for the design of new materials structures and thus new functionalities Previous reports reported that E is highly correlated with certain predictors which are simple functions of certain molecular properties [4]. These molecular properties are calculated using Density Functional Theory (DFT). Thus, E for molecules can be predicted by their own computational methods before testing their electrical and chemical behavior in experiments.
In this research, density functional theory (DFT) has been developed as reliable and effective approach for the computer simulation of molecular structure and also to measure the energy for chemical reaction. DFT is also can be used to search the efficiency and acuracy of the molecular properties, such as electronegativity, chemical potential and affinities [5]. The molecular properties of can be accurately described and calculated by manipulating the electron density and its fundamental quantity.
2. Research Methodology
2.1. Preparation of Molecular Gases
Preparation of molecular gases i.e. O2, N2¸ H2, CO and CO2 are beginned with skecthed of each molecular gases using ChemBioDraw ultra 13.0 for then it should save it in mdl. mol format. for then imported it into the Gaussian software.Finally, complete content and organizational editing before formatting. Please take note of the following items when proofreading spelling and grammar.
2.2. Density Functional Theory (DFT)
The initial structure of each gas O2, N2, H2, CO and CO2 are needed to obtain, density functional theory (DFT) is urgently needed to optimize the stability of these gases. DFT was runned using Gaussian 09 software with 6-311G/B3YLP was selected as basis set. Basis set is shape of atomic orbital that explained using wave function. TD-SCF was used as method to perform the Gaussian [6]. Stability of the calculation was explored the stability of the wavefunction computed for a molecular system. The stability of SCF solution for unknown systems should always be tested. The consideration for stability of unknown systems should be apply and may be tested in calculation using DFT methods as well.
3. Results and Discussion
Single particle are described about molecules or atom or also described how the chemical bonding between each atom to make a molecule. In this study, density functional theory (DFT) was applied to check the stability of gases and also to study shock induced process because they involve spatial and temporal scales that go beyond those attainable at the atomistic level. The stability of each gas such as N2¸O2, CO, CO2¸ and H2. base on the DFT calcution we will know charge distribution and excitation energy [7]. DFT calculation provides the energy of ionization potential for these gas elements.
Based on DFT simulation for these gases N2, O2, H2, CO and CO2, these gases are looks stable with the excititation energy of 0.56334 eV, 0.56334 eV, 0.49436 eV, 0.52185 eV, 0.53651 eV, respectively. Plate testing model (anode and cathode electrodes) was used for high voltage for then it can be simulated the excitation energy using the equation 1 (eq. 1) with d = 0.01 m. The results of the calculation of excitation energis is presented in Table 1.
W = F.d (1)
Where, W is excitation energy (eV) and F is Coulomb force on electrical charges (N)
F = E . e (2)
E is electric Field/ electrical strength (V/m) and e is electron
Initially, the DFT for each gas was evaluated using Gaussian 09 software package. Instead of DFT, it also can predict the spectrum UV-Vis for each gas. Based on spectrum UV-Vis, the information about energy excitation will be informed [8].
3.1. Nitrogen
The gas N2 is seems to be stable at the wave length 89.17 nm, based on DFT calculation for the best pose selected with the following results; energy of excitation of 89.17 eV, energy for the ionization process of 108.98 au from orbital eight of -0.13492 to orbital seven of -0.42842. The ionization process of molecule N2 is depicted in Figure 1.
Molecule consisted of atoms, atoms consisted of electrons, proton and neutron. Excitation of each electron in orbital atoms can cause the ionization process. The ionization process is stable if the range energy between two orbital less than 0.9 eV [9]. For gas N2, there are two electrons was jumped from orbital eight (4p) into orbital seven (4s) with the range energy of 0.56334 eV. It is indicated that gas N2 is stable.
Table 1. Gases excitation energy
| No | Gases | W (eV) | E (V/m) |
| 1 | Molecule N2 | 0.56334 | 56.334 |
| 2 | Molecule O2 | 0.56334 | 56.334 |
| 3 | Molecule H2 | 0.49436 | 49.436 |
| 4 | Molecule CO | 0.52185 | 52.185 |
There are some reasons to make the frontier orbital become so significant, they are: method for calculating the ionization energy and Koopman’s theory. Based on Homo-Lumo calculation on our previous paper [10], it is provided the energy of ionization potential for gas elements. Furthermore, DFT calculation is needed to analyze much more about the orbital for basic gas.
Each molecule has orbital or molecular orbital (MO), it can be used to represent the regions in a molecule where an electron occupying that orbital is likely to be found. Molecular orbitals also can be used to predict the location of an electron in an atom. A molecular orbital can specify the electron configuration of a molecule: the spatial distribution and energy of one (or one pair of) electron(s). The molecule will stabilize if the electrons in the last orbitals have their paired up [11].
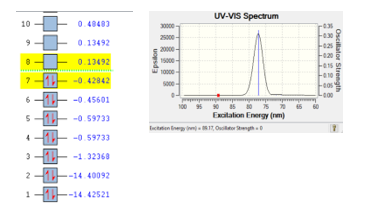 Figure 1: Ionization process of gas N2
Figure 1: Ionization process of gas N2
3.2. Oxygen
Generally, DFT can be defined as method for computational quantum mechanical modelling used in physic, chemistry or material science for investigating the properties of electron in a system, hence the name density functional theory comes from the use of functionals of the electron density [12].
Like gas N2, ionization of molecule O2 is looks stable since the excitation energy of -395143 eV with the wave length of 395143 nm, the best pose selected with the following results; energy for the ionization process of 108.98 au from orbital nine of 0.01523 to orbital eight of -0.05553. The ionization process of molecule O2 is presented in Figure 2. Stability of gas O2 is shown with there are two electrons was jumped from orbital nine (4d) into orbital eight (4p) with the range energy of 0.07053 eV.
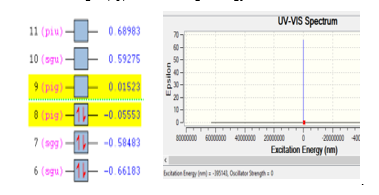 Figure 2: Ionization process of gas O2
Figure 2: Ionization process of gas O2
3.3. Hydrogen
The stability of molecule H2 is also checked using the same way like molecule O2 and N2. Base on DFT calculation, it come out with the excitation energy of 88.8 eV at the wave length 88.8 nm. Two electrons were jumped from orbital two into orbital one with the energy of 0.49436 eV. Figure 3 is presented the ionization process of molecule H2.
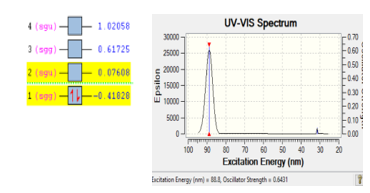 Figure 3: Ionization process of gas H2
Figure 3: Ionization process of gas H2
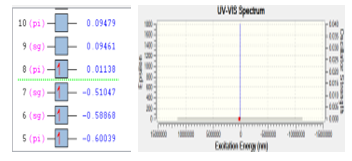 Figure 4: Ionization process of gas CO
Figure 4: Ionization process of gas CO
3.4. Carbon Monoxide
Stability of molecule CO was checked using DFT. The first principles of DFT is to carried out the structural stability and also the electronic properties of a molecule. Base on DFT calculation, gas CO has the excitation energy of 167782 eV. Only one electron was excited from orbital eight into orbital seven with the energy of 0.52185 eV. The process of electron excitation for molecule CO is depicted in Figure 4.
3.5. Carbon Dioxide
DFT determined the stability of molecule CO2. Base on the DFT simulation, gas molecule CO2 is also looks stable with the excitation energy of 96.13 eV. There are two electrons were excited from orbital eleven into orbital ten with the energy of 0.53651 eV. Figure 5 is presented the ionization process of molecule CO2.
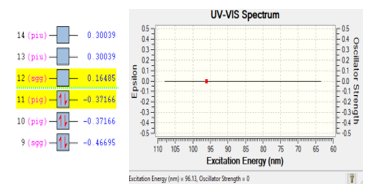 Figure 5: Ionization process of gas CO2
Figure 5: Ionization process of gas CO2
4. Conclusion
There are several conclusions that can be concluded base on this research:
- Density functional theory (DFT) has been developed as reliable and effective approach for the computer simulation of molecular structure.
- The ionization process is stable if the range energy between two orbital less than 0.9 eV.
- Gas molecule N2, O2, H2, CO and CO2 is stable with the excititation energy of 0.56334 eV, 0.56334 eV, 0.49436 eV, 0.52185 eV, 0.53651 eV
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
We thank to Lembaga Penelitian dan Pengabdian Kepada Masyarakat (LPPM) Universitas Riau for the financial support through Penelitian DIPA UNRI percepatan inovasi 2019, contract Number :1026/UN.19.5.1.3/PT.01.03/2019
- L. B. Affoue, B. A. Amon, D. Y.. Kicho, G. K. Mamadou, K. Soleymane, K. Mawa, R. N. Boka, S. B. El-Hadj, “Theoretical study by density functional theory method (DFT) of stability,tautomerism, reactivity and prediction of acidity of quinolein-4-one derivatives” Computational Chemistry. 6, 57-70, 2018. https://doi.org/10.4236/cc.2018.63005
- G. Biasutti,’Neue Isoliergase für gekapselte Hochspannungsanlagen”. Ph.D. Thesis, Diss. ETH Nr. 7683, 1985
- R. E. Robson, P. B. Nicoletopoulos, R. D. Li, “Kinetic theoretical and fluid modelling of plasmas and swarms: the big picture” Plasma Sources Science and Technology, 17, 024020, 2008. https://doi:10.1088/0963-0252/17/2/024020
- M. Rabie, D. A Dahl, S. M. Donald, M. C. Reiher, M. Franck,. “Predictors for Gases of High Electric Strength”. IEEE Transactions on Dielectrics and Electrical Insulation, 20 (3), 856–863, 2013. https://doi: 10.1109/TDEI.2013.6518955
- M. S. Jorge, “An introduction to density functional theory in chemistry” 2018. Theoretical and computational chemistry, 2, 1-27, 2018. https://doi.org/10.1016/S1380-7323(05)80031-7
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, “Gaussian 03, Revision A.1” Gaussian Inc., Pittsburgh, 2003
- A. Nathan, M. Guy, “Density Functional Theory — an introduction” American Journal of Physics. 68 (1), 69-79, 2000. https://doi.org/10.1119/1.19375
- K. Erick, L.Vladimir, “Study of gas-phase O–H bond dissociation enthalpies and ionization potentials of substituted phenols – Applicability of ab initio and DFT/B3LYP methods” Chemical Physics, 330, 515–525, 2006. https://doi.org/10.1016/j.chemphys.2006.09.026
- J. Ahmad, J. Li, M. Hussain, M. T. Riaz, M. A. Akbar, and M. Z. Khan, “Study of Physio-chemical Properties of POSS/Mineral Oil based Nanofluids,” in 2018 International Conference on Power Generation Systems and Renewable Energy Technologies (PGSRET), 2018, pp. 1–5. https://doi.org/10.1109/PGSRET.2018.8685972
- M. Kurt, T. R. Sertbakan, M. Ozduran, “An Experimental and Theoretical Study of Molecular Structure and Vibrational Spectra of 3- and 4-Pyridineboronic Acid Molecules by Density Functional Theory Calculations”. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 70, 664-673, 2008. https://doi.org/10.1016/j.saa.2007.08.019
- Q. Hai, L. Wenhao, W. Xiaoxing, C. Wei, D. Jun, H2S and SO2 adsorption on Pt-MoS2 adsorbent for partial discharge elimination: A DFT study. Results in Physics, 12, 107-112. 2019. https://doi.org/10.1016/j.rinp.2018.11.035
- Z. Yubo, W. James, X. F. Bing, S. Jianwei, “Subtlety of TiO2 phase stability: Reliability of the density functional theory predictions and persistence of the self-interaction error” The Journal of Chemical Physics. 150 (01), 014105. 2019. https://doi.org/10.1063/1.5055623
Citations by Dimensions
Citations by PlumX
Google Scholar
Scopus
Crossref Citations
- Mudar Ahmed Abdulsattar, Nooruldeen Mudher Almaroof, Hashim Rashid Jabbar, "Interaction thermodynamics of human hemoglobin with environmental and toxic gases: A density functional theory study." Journal of Physics: Conference Series, vol. 1963, no. 1, pp. 012132, 2021.
No. of Downloads Per Month
No. of Downloads Per Country